Advanced computing at UNC Charlotte indicates current antibodies effective against newly emergent SARS-CoV-2 XBB.1.5

A team at UNC Charlotte’s Center for Computational Intelligence to Predict Health and Environmental Risks (CIPHER) and Tuple, a Charlotte-based genomics consulting firm, has used artificial intelligence to rapidly assess the public health implications of the newly emergent SARS-CoV-2 XBB.1.5 variant. Results from simulations run by the team indicate the antibodies currently in our arsenal are effective to neutralize SARS-CoV-2 XBB.1.5.
The variant has recently increased its share of COVID-19 infections caused in the United States, creating concern about a potential health care crisis. However, increased transmission ability of the variant does not mean it is not treatable.
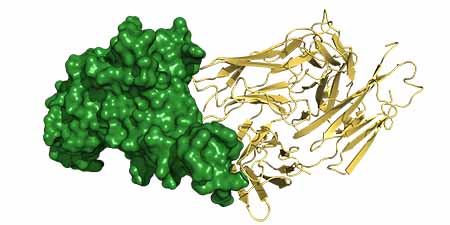 |
|
Therapeutic antibody (Tixagevimab; AZD8895, shown in gold) docked against the new viral variant (SARS-CoV-2 XBB.1.5 receptor binding domain, shown in green) |
“The CIPHER and Tuple team have tied several technologies together to provide rapid health intelligence, which is a dose of good news. It is becoming more clear that with new sciences and hard work we can address emergent diseases,” said Daniel Janies, the co-director of the CIPHER center and Carol Grotnes Belk Distinguished Professor of Bioinformatics and Genomics.
The team used machine learning applications to simulate protein structure and antibody binding. That way, they tested a variety of antibodies including those induced by the Omicron booster vaccine and others used in therapies.
These results are in contrast to those found by the team when the original Omicron variant emerged over a year ago. In 2021, the team correctly predicted the original vaccine and antibody arsenal had reduced efficacy against Omicron.
As this team works in computational simulations on digital representations of the variants and the antibodies, the work is very quick. The team uses protein structures from the program AlphaFold2, then a program called HADDOCK to dock the antibodies to the variants’ receptor binding domain in the Spike protein of SARS-CoV-2 XBB.1.5 and related viruses. The assessment is based on physics models to test the quality of docking and predict maintained or weakened efficacy of the antibodies against the viruses.
The computational work precedes, by weeks, the structural and binding experiments that must be done in a traditional chemistry laboratory. CIPHER researchers leverage new tools and UNC Charlotte’s advanced computing capabilities to provide a valuable early indication of what health officials can expect from the rise of a new variant as well as the therapeutics and vaccines that are effective in response.
A major investment by the N.C. General Assembly helped to fund this research and the CIPHER center.
The full paper on XBB.1.5 and related viruses can be found here:
The peer reviewed Omicron paper using the same methodology can be found here:
The authors include:
Colby T. Ford (1, 2, 6)
Shirish Yasa (3, 4, 6)
Denis Jacob Machado (3, 4 ,6)
Richard Allen White III (3, 4, 5, 6)
Daniel Janies (3, 4, 6)
1 Tuple, LLC, Charlotte, NC, USA
2 School of Data Science, University of North Carolina at Charlotte, Charlotte, NC, USA
3 Department of Bioinformatics and Genomics, University of North Carolina at Charlotte, Charlotte, NC, USA
4 Center for Computational Intelligence to Predict Health and Environmental Risks (CIPHER), University of North Carolina at Charlotte, Charlotte, NC, USA
5 North Carolina Research Campus (NCRC), University of North Carolina at Charlotte, Kannapolis, NC, USA
6 College of Computing and Informatics, University of North Carolina at Charlotte, Charlotte, NC, USA
Image, top, Computational workflow of the predictive analyses from sequence, to protein structure generation, to protein-antibody docking. The red circles indicate the areas of structural interaction between the receptor binding domain (RBD) of the viral protein and the fragment antigen binding (Fab) region of the antibodies.